

Overview
With the rapid development of sequencing technologies, especially the maturation of long-read sequencing technologies (Pacific Biosciences and Oxford Nanopore sequencing), the number and quality of published genome assemblies have significantly increased. More and more genome assemblies are being published, widely promoting the development of biological research. The reliability of genome assemblies is crucial for downstream functional genomics studies, but assemblies are always prone to errors, and the types of misassemblies can include insertions, deletions, inversions, duplicate folding, and duplicate expansion. Therefore, assessing the quality of genome assembly is a necessary and important process.
What difficult do we face?
Assessment of genome assembly quality is a challenging and complex task. The difficulty mainly stems from the fact that we never know the true genome sequence. Therefore, a combination of strategies to assess the assembly quality is a common and effective solution until the sequence data and the generated assemblies are able to reach the reference quality on a regular basis. The assessment of genome assembly quality is usually based on three aspects: continuity, completeness, and correctness, often referred to as the 3C principles.
However, these 3C principles are actually contradictory; higher continuity means more ambiguous nodes to be dealt with, which can lead to an increase in the overall error rate, and in order to ensure complete correctness, then it leads to a very fragmented continuity. In addition, these 3C principles are also more qualitative, and we need more quantitative numerical measures. Currently the most commonly used measures of genome assembly quality address only two of the 3C, with the more commonly used metrics being N50 and BUSCO/CEGMA.
The Genome Assembly Evaluation Pipeline (GAEP)
Many different metrics or methods can be used to assess assembly continuity, completeness, and correctness. Genome projects often rely on the choice of these metrics or methods to assess the three aspects of the assembly. However, the choices may vary from project to project, and even for the same metrics, using different pipelines or parameters may lead to different results, which poses a challenge in comparing different assemblies horizontally.
In addition, evaluating genomes using different methods can be a cumbersome task, as it usually requires installing multiple software packages and debugging various parameters. This situation results in the fact that not all published genomes are comprehensively evaluated, leading to a lack of confidence in their quality. Three tools, the Genome Assembly Evaluation Pipeline (GAEP), GenomeQC, and QUAST, address these challenges. With a range of methods and tools, based on different types of orthologous data, researchers can better assess the quality and accuracy of genome assemblies, making them more valuable for biological applications and interpretation.
Assessing the Quality of Genome Assembly with the 3Cs
Continuity Assessment
Continuity, which largely represents the effective extension of the assembled sequence, is designed to measure the uninterrupted extension of genomic regions and is a direct measure of assembly effectiveness. Nx metrics are still the primary measure, e.g., the N50 value indicates the length of the shortest overlapping cluster representing 50% of the genome. With advances in sequencing technology, an overlapping group N50 of more than 1 Mb, especially in long-read sequencing assemblies, is often considered satisfactory. In addition to Nx metrics, the number of gaps and overlap clusters are important parameters that reflect potential breaks in the assembly.
Integrity Assessment
Integrity aims to assess the inclusion of the entire original sequence in the assembly as far as possible. The main methods are:
Flow cytometry
Completeness is estimated by comparing the length of the assembled genome to the estimated genome size using flow cytometry.
K-mer spectra and mapping ratios
Compare k-mer profiles obtained from assemblies with k-mer profiles from high-precision sequencing reads, such as Next Generation Sequencing (NGS) reads. The ratio of shared k-mers to total k-mers from the reads can indicate the integrity of the assembly. In addition, mapping whole-genome sequencing reads to assemblies, the mapping ratio can indicate assembly completeness.
Correctness Assessment
Correctness can be defined as the accuracy of each base pair in an assembly and is most often measured as the agreement of the assembly with the gold standard reference. It involves evaluating both base-level and structural-level accuracy.
Base-level assessment
A popular approach is to map NGS reads to assemblies. By doing this, it can identify pure fit variants, providing insight into base level correctness. However, challenges remain, including non-specific alignment of duplicate regions or sequencing imbalances.
K-mer analysis
To circumvent these problems, k-mer spectral comparisons between reads and assemblies have become an effective alternative. Such methods provide a more direct measurement of baseline accuracy by eliminating mapping-induced differences. However, researchers must remain vigilant, as regions with heterogeneity or duplicity can still impact the results.
Structural-level assessment
Delving deeper, the structural accuracy of assemblies goes beyond individual bases to focus on larger genomic configurations. Reference-based tools (e.g., QUAST) can be used as benchmarks to assess structural correctness by identifying structural variants in reference genome assemblies. However, a limitation of this approach is that the reference genome is not always available and, more importantly, it is unable to distinguish between actual genetic variation and misassembly. Another approach is based on whole genome sequencing reads, which relies on identifying breakpoints from the process of mapping reads to assemblies. Reads can be short reads from NGS technology and long reads from third-generation sequencing technology. The final method involves manually checking the assembly, supplemented with the reference genome, Hi-C, or Bionano data.
In addition, there are other evaluation strategies based on conserved gene sets, such as BUSCO and CEGMA. these methods can effectively assess the status of conserved genes in the assembly and thus infer the assembly effect. In addition, researchers widely use the LTR assembly index (LAI) to evaluate plant genomes by calculating LTR completeness to assess assembly quality.
Tools for Assessing the Quality of Genome Assembly
Genome Assembly Evaluation Process (GAEP)
GAEP is a comprehensive tool for assessing the continuity, accuracy, completeness, and redundancy of assembled genome sequences using NGS data, long-read data, and transcriptome data. Specifically, GAEP utilizes these data sources to evaluate assemblies. The basic statistics module can automatically generate a series of evaluation metrics, such as total length, contig/scaffold number, gap-free length, gap number, Nx metrics, etc. GAEP also integrates a BUSCO processing script to evaluate the integrity of homologous genes.
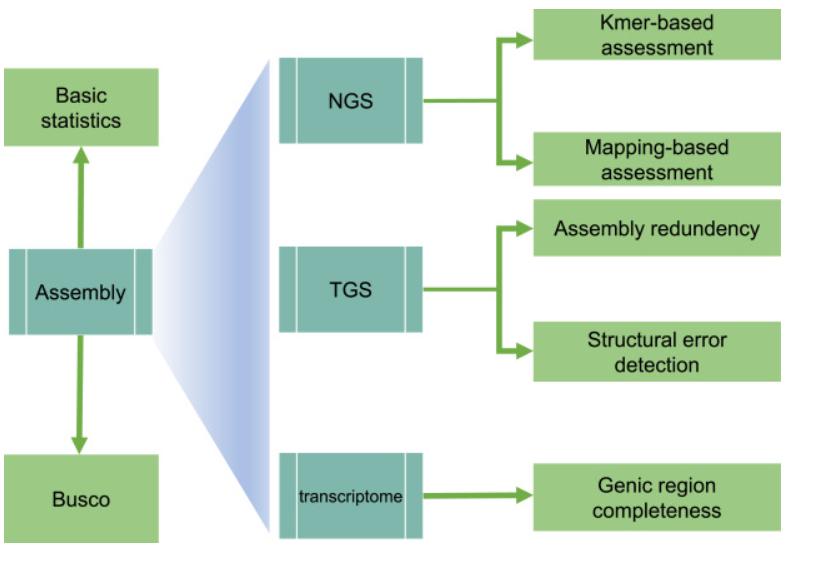
Overview of the GAEP pipeline. (Zhang et al., 2023)
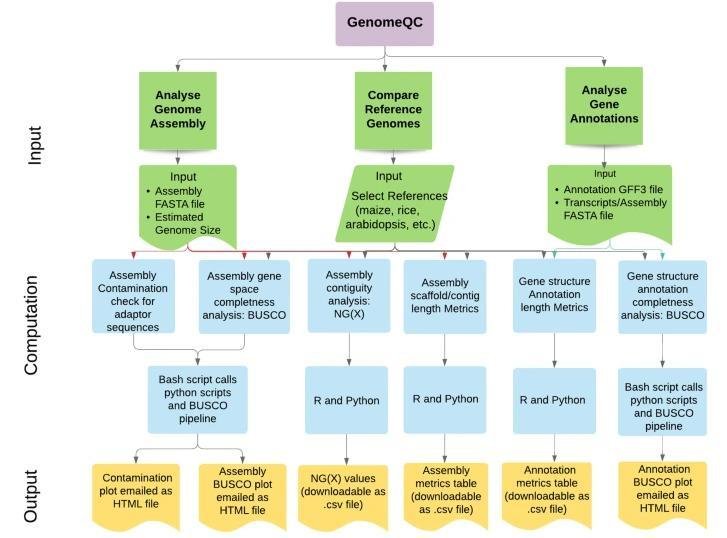
GenomeQC
GenomeQC is a robust, comprehensive tool offering diverse quantitative metrics to evaluate genome assemblies and their annotations. One of the strengths of GenomeQC is its ability to act as an interactive web framework. This enhances usability, allowing researchers to swiftly compare assemblies and benchmark them against gold standard references. In terms of assembly continuity, GenomeQC emphasizes metrics such as N50/NG50 and L50/LG50, which provide a consistent measure of assembly continuity.
For completeness, GenomeQC utilizes an innovative strategy. Tools like BUSCO are essential, offering quantitative measures of genomic integrity by assessing expected gene content.
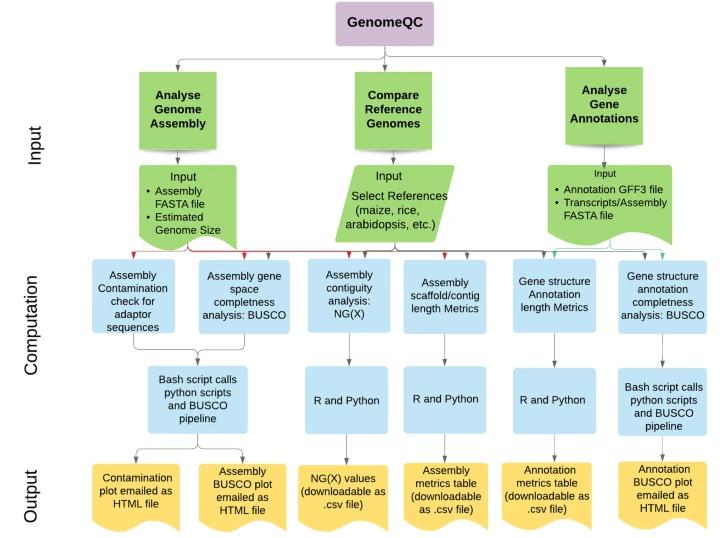
Workflow of the GenomeQC web application. (Manchanda et al., 2020)
QUAST
QUAST became an indispensable tool in the field of genome assembly quality assessment. The core objective of QUAST is to provide a comprehensive and integrated approach to assessing genome assembly. The tool is versatile and capable of assessing assemblies with or without a reference genome. Unlike traditional methods, QUAST provides a balanced set of metrics to ensure a broad and sensitive assessment. It focuses on integrating diverse metrics to thoroughly assess the continuity, completeness, and accuracy of the genome assembly. The tool is for prioritizing usability and efficiency, integrating parallel processing to optimize evaluation.
A notable feature of QUAST is its adaptability. QUAST efficiently evaluates assemblies of previously unsequenced species, addressing the limitations of tools that depend on completed genomes. This versatility ensures that it remains the tool of choice for bioinformaticians in all fields.
Read More: single-nucleotide-variants-whole-analysis
References
- Zhang, Yong, Hong-Wei Lu, and Jue Ruan. “GAEP: a comprehensive genome assembly evaluating pipeline.” Journal of Genetics and Genomics (2023).
- Manchanda, Nancy, et al. “GenomeQC: a quality assessment tool for genome assemblies and gene structure annotations.” BMC genomics 21.1 (2020): 1-9.
More Stories
PCD Pharma Franchise in Uttar Pradesh – A Lucrative Business
Why Skin Care Mistakes Can Lead to Long-Term Damage
Top PCD Pharma Companies in Karnataka